Today I want to talk about polymer-drug conjugates. What’s the idea behind such conjugates? What are the advantages and challenges? And are there any polymer-drug conjugates in clinical use?
I became interested in this topic during my PhD, as I was developing a polymer carrier material for ocular use. Since my carrier should be water-soluble, conjugation was the best way to incorporate drugs into the formulation. The concept of polymer-drug conjugates had already been described in the 1970s,[1] but hadn’t really been pursued in ocular drug delivery. Here, I will highlight some advantages of this technology and if you are interested to learn more, you can read my PhD thesis here.
Terminology
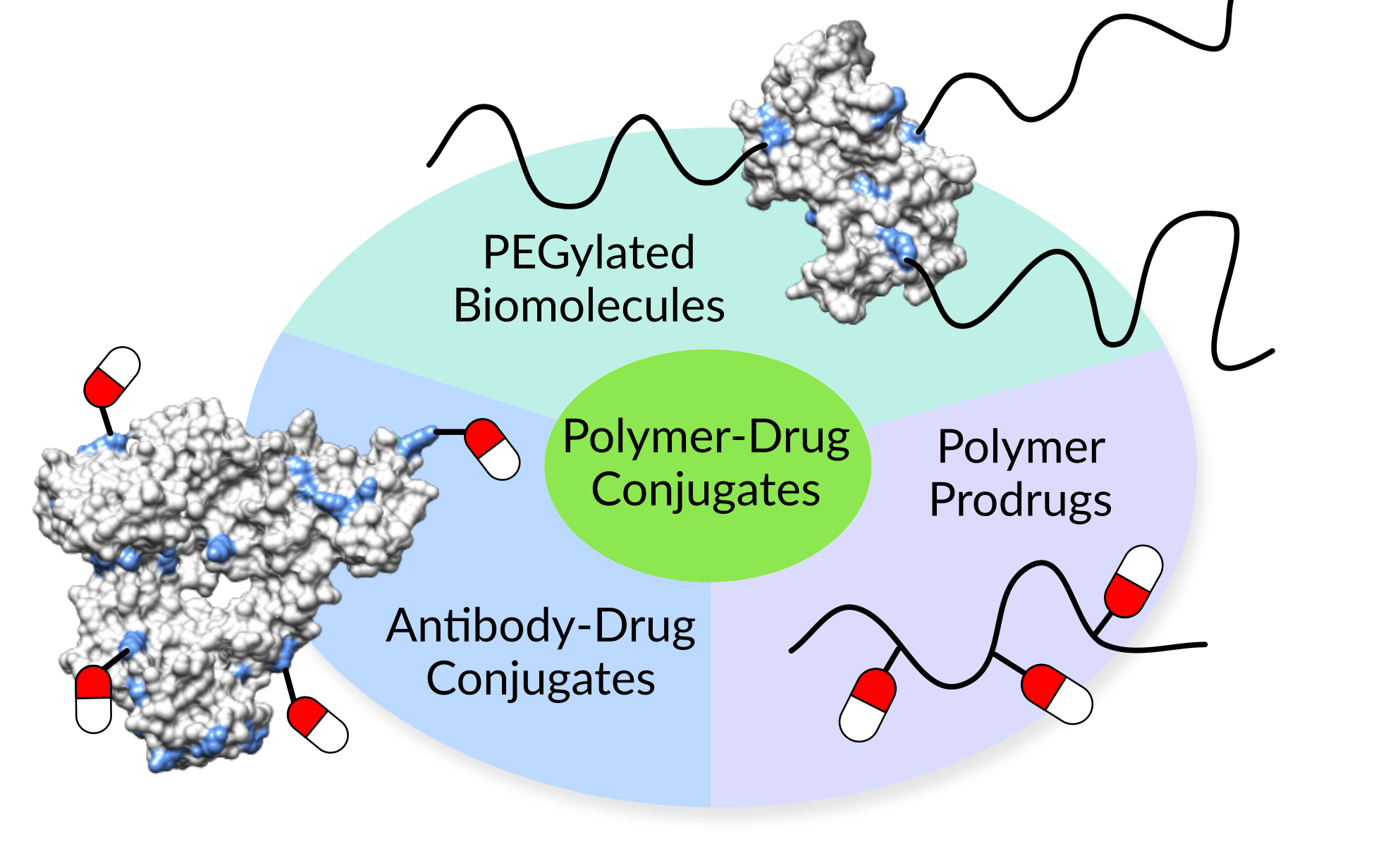
The word drug, in this context, describes any entity with a therapeutic effect. That can be small molecules, peptides, proteins (e.g. antibodies or enzymes) or even RNA/DNA strands. The polymer can be of natural or synthetic origin and feature any architecture, from linear to branched to crosslinked. Most importantly, there is a covalent bond between the drug and the polymer, making it a conjugate. Now, we can distinguish two types of drug conjugates depending on their mode of action:
- Polymer-drug conjugates, where the drug is active despite being coupled to a macromolecular carrier.
- Polymer prodrugs, where the drug is inactive until it is released in its native form.
Benefits of polymer-drug conjugates
In both cases, conjugation of drugs to polymers enhances their solubility and stability in body fluids. It also reduces their toxic side effects in healthy tissues. The covalent bonds allow for incorporation of higher drug loads compared to physically entrapped formulations. Carefully designed linkages between drugs and polymers make it possible to control the release profile. Additional functional groups of the polymer can be used to attach ligands and target the drugs to specific cells and tissues.
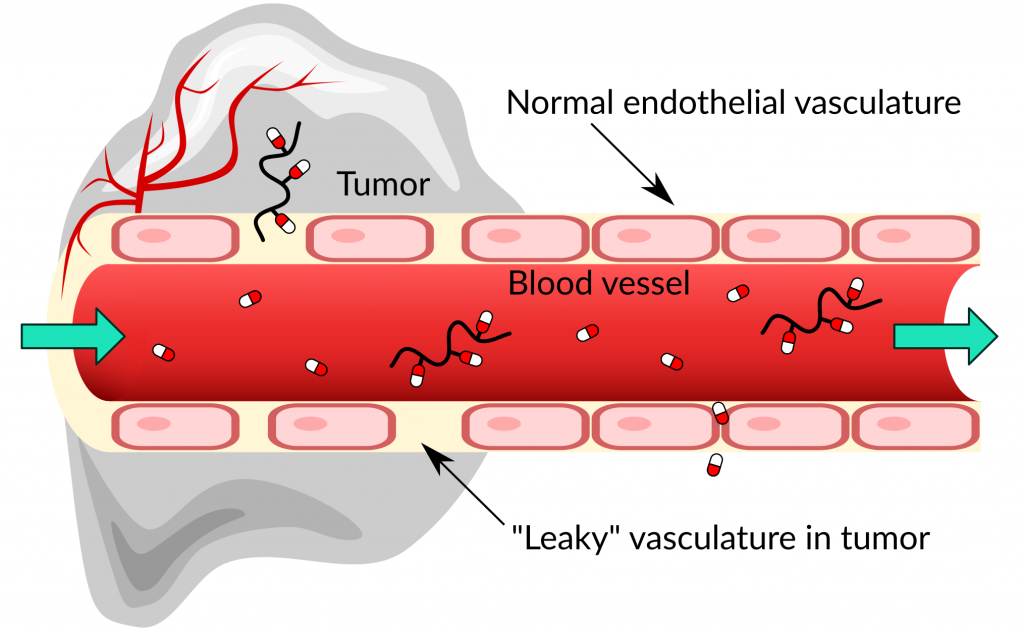
The main advantage of the conjugate compared to the individual drug is its significantly enhanced residence time in the body. This is a result of the high molecular weight of the polymer. For example, take a small molecular drug injected into the blood stream. Because of its small size, the drug can squeeze through the narrow gaps between the cells in the blood vessel walls and penetrate into all tissues of the body (Figure 1). The drugs will also circulate with the blood through the kidneys, where they are filtered out. A large polymer on the other hand is unable to escape from healthy blood vessels and it evades renal excretion. The threshold of kidney filtration is about 30 kDa.[2] Thus, larger polymer-drug conjugates circulate longer in the body, prolonging the drug action and/or dosing intervals.
EPR effect & Cellular targets
The inability of polymers to escape healthy blood vessels can be exploited to passively target cancerous tumors. The blood vessels in some tumors are leaky and allow macromolecules to infiltrate the tissue. At the same time, lymphatic drainage from the tumors is inefficient, so that the polymers will accumulate there. This process is called “enhanced permeation and retention” (EPR) effect.[3]
At the site of action, the drug must be able to interact or bind with its target in order to exercise its therapeutic effect. The majority (60 %) of known drug targets are proteins located at the cell surface.[4] Other targets are intracellular proteins, ribosomes or DNA. Depending on whether the drug is active or inactive in its coupled form and on the target location, the polymer-drug conjugate may need to release the drug (Figure 2).
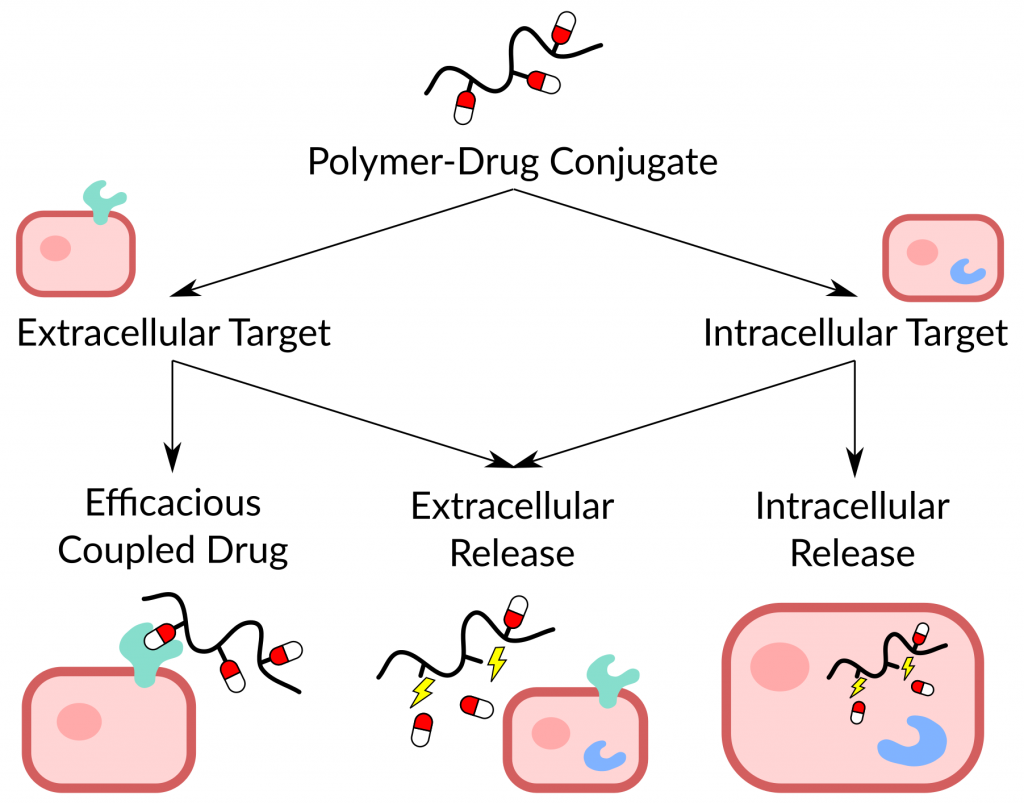
If the drug is active and the target is accessible from the extracellular space, a therapeutic effect can be elicited. In case of an inactive prodrug and/or when the macromolecular carrier cannot enter into the target cell, drug release must take place outside the cell. Is the drug target inside the cell and the polymer carrier can enter, the drug might still need to be released intracellularly. Luckily, there is a different (bio)chemical environment inside and outside the cells. We can use this difference to design linkages between the polymers and drugs that break under specific conditions. Thereby we can control the site of drug release.
Linkages
The linkages between polymers and drugs can be stable, or they can be chemically or enzymatically cleavable. Chemical triggers for the breakage of bonds are pH, oxidative/reductive environments or the presence of reactive groups. For example, disulfide bonds are stable in the blood and extracellular fluid, as both are slightly oxidative. But they break in the intracellular environment and in some tumor tissues, because of a higher concentration of reducing agent glutathione.[5] They also undergo reshuffling in the presence of free thiols. Ester bonds hydrolyze at neutral pH in blood. Their rate of cleavage can be tuned by the nature of neighboring groups. Hydrazones and acetals remain stable until they reach the acidic intracellular compartment. The pH drop from 7.4 to around 6.5 – 4.5 in cell organelles is a reliable trigger that is constant across patients.[6]
Very high target site specificity can be achieved with peptide linkers, which are cleaved by specific enzymes, so-called proteases. Different enzymes exist exclusively in the intra- or extracellular space and can be targeted with carefully crafted peptide sequences.[7] Steric hindrance plays a bigger role in the design of these conjugates. Polymers are bulky and if the peptide linker is too close to the polymer backbone, enzymes may not be able to access it. Also, the concentration or even presence of enzymes may differ across patients and disease subtypes.
Examples of polymer-drug conjugates
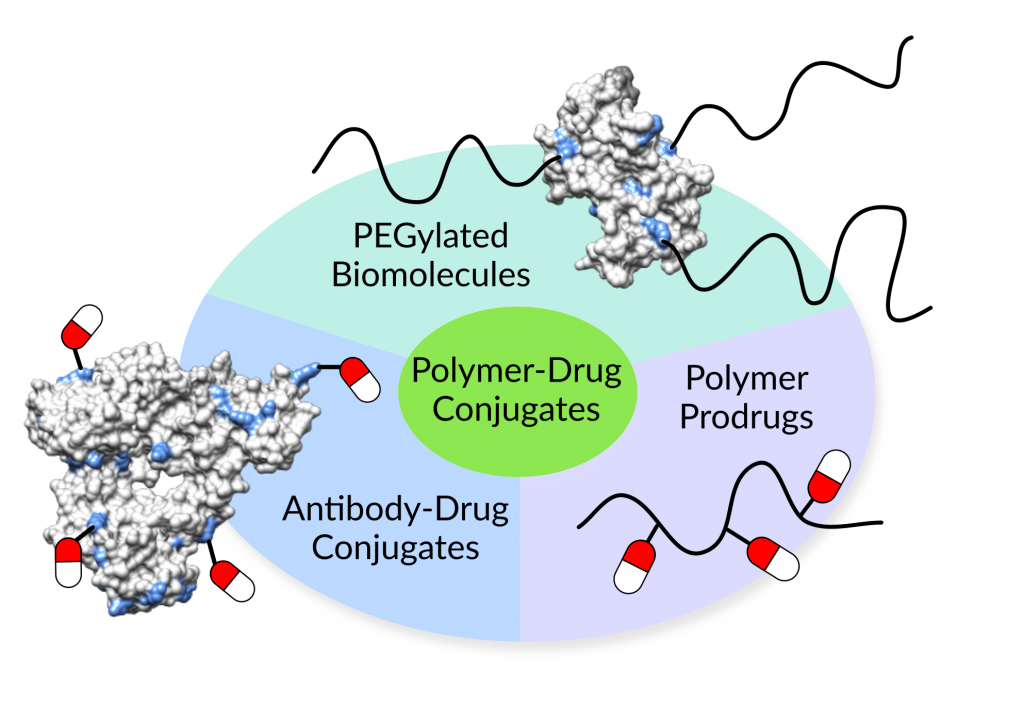
Now on to some examples of polymer-drug conjugates that have made their way to the market. The American Food and Drug Administration (FDA) has approved a number of PEGylated biopharmaceuticals.[8] Those are proteins, antibodies or aptamers that carry one or more polyethylene glycol (PEG) polymers. For instance, Macugen® (Valeant Pharmaceuticals International) is a PEGylated single-strand of nucleic acid, a so-called aptamer. It binds to and blocks one form of the vascular endothelial growth factor (VEGF). VEGF is responsible for the growth of abnormal blood vessel in wet age-related macular degeneration. Other examples include immunodeficiency (Adagen®, Leadiant Biosciences), acromegaly (Somavert®, Pfizer), and hepatitis C (PEG-Intron®, Merck Sharp & Dohme) therapies, among others. PEGylation improves the stability of biopharmaceuticals and enhances their circulation time in the body.
Antibody-drug conjugates are another class of conjugates that receive a lot of attention right now. There are four currently approved products, all acting against cancer.[9] Kadcyla® (Genentech/Roche) was developed for patients with previously treated metastatic breast cancer.[10] Adcetris® (Seattle Genetics and Millennium Pharmaceuticals/Takeda Oncology) is active against classical Hodgkin lymphoma and T-cell lymphoma.[11] And Wyeth Pharmaceuticals/Pfizer released two new leukemia therapeutics (Besponsa™ & Mylotarg™) last year. All of them feature a similar mode of action. The antibody first binds to receptors that are specific to cancer cells and overexpressed on their surface. Thereafter, the conjugate is taken up into the cells and releases its drugs.
Translational challenges
In contrast to the number of successful biopharmaceuticals, polymer-drug conjugates based on synthetic or natural polymers have not found their way to the market, yet. There are a number of ongoing clinical trials for polymer conjugated cancer drugs, though.[12] Besides PEG, many of these conjugates comprise the synthetic polymer of N-(2-hydroxypropyl) methacrylamide (HPMA). Further candidates include naturally occurring polysaccharides, such as hyaluronic acid, dextran, or cyclodextrin, as well as semi-synthetic polymers made of the amino acid α,L-glutamic acid.
While polysaccharides and amino acid-based polymers are degradable, synthetic polymers are often not. They can accumulate in the body if their molecular weight is too high, unless they are carefully designed to include breaking points. Despite advances in controlled polymerization techniques, polymer-drug conjugates still feature significantly broad molecular weight distributions. Conjugate variability is one major challenge in passing the stringent regulatory approval process. Also, the synthetic procedures to produce polymer-drug conjugates are often extremely complex and hinder the scale-up and commercial viability of the products.
Nevertheless, there is huge potential in polymer-drug conjugates. With their adjustable physicochemical properties and large number of attachment sites, polymers are the ideal carriers for e.g. combination drug therapy. A closer collaboration of industrial and academic scientists could improve the design of scalable materials. Finally, new pre-clinical and clinical models that test the behavior and fate of the conjugates in the body will allow us to optimize the architecture, linkages and targeting moieties of our constructs.
References
[1] Helmut Ringsdorf, “Structure and properties of pharmacologically active polymers”, Journal of Polymer Science: Polymer Symposia, 1975, 51 (1), 135-153. doi:10.1002/polc.5070510111.
[2] Camilla Frich Riber & Alexander N. Zelikin, “Recent advances in macromolecular prodrugs”, Current Opinion in Colloid & Interface Science, 2017, 31, 1–9. doi:10.1016/j.cocis.2017.06.002.
[3] Jun Fang et al., “The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect”, Advanced Drug Delivery Reviews, 2011, 63 (3), 136-151. doi:10.1016/j.addr.2010.04.009.
[4] John P. Overington et al., “How many drug targets are there?” Nature Reviews Drug Discovery, 2006, 5, 993–996. https://www.nature.com/articles/nrd2199.
[5] Ajit Joseph M. D’Souza & Elizabeth M. Topp, “Release from polymeric prodrugs: linkages and their degradation”, Journal of Pharmaceutical Sciences, 2004, 93 (8), 1962–1979. doi:10.1002/jps.20096.
[6] Yong-Bo Hu et al., “The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration”, Translational Neurodegeneration, 2015, 4 (18). doi:10.1186/s40035-015-0041-1.
[7] Rubi Mahato et al., “Prodrugs for improving tumor targetability and efficiency”, Advanced Drug Delivery Reviews 2011, 63 (8), 659–670. doi:10.1016/j.addr.2011.02.002.
[8] Peter L. Turecek et al., “PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs”, Journal of Pharmaceutical Sciences, 2016, 105 (2), 460-475. doi:10.1016/j.xphs.2015.11.015.
[9] ACD Review News, 20.01.2018, accessed 17.11.2018.
[10] Kadcyla® website, accessed 17.11.2018.
[11] Adcetris® website, accessed 17.11.2018.
[12] Douglas R. Vogus et al., “A review on engineering polymer drug conjugates to improve combination chemotherapy”, Current Opinion in Colloid & Interface Science, 2017, 31, 75–85. doi:10.1016/j.cocis.2017.08.002.
well written 🙂